Xevo G2-S QTof and TransOmics: LC/MS differential omics analysis system for proteomics, metabolomics and lipidomics
Ian Edwards, Jayne Kirk, and Joanne Williams
Waters Corporation (Manchester, UK)
Application advantage
â– Simplified workflow, validation and data analysis â– Designed for large-scale metabolomics and proteomics data sets â– Integrated omics platform for a wide range of comprehensive qualitative and quantitative analyses
Waters <br> solutions include genomics research platform TransOmics â„¢ informatics software solutions
ACQUITY UPLC® I-Class System
nanoACQUITY UPLC® System
Xevo® G2-S QTof
TransOmics informatics software
MassPREPTM Protein Enzymatic Standard
Key words
Omics, metabolomics, lipidomics, proteomics, MS E , principal component analysis, label-free LC/MS
About <br> recent years, including advances in LC-MS-based metabolomics, and proteomics instruments lipids such as genomics technologies to achieve a high throughput manner on a variety of abundance biomolecules quantitatively Monitor to determine their changes in different biological states.
Our ultimate goal is to improve understanding of biological processes, thereby improving the efficacy of the disease, developing drugs more efficiently or maintaining the optimal agricultural environment for crop growth, while minimizing infection and other side effects. In this regard, the results of different analytical disciplines can provide orthogonal perspectives that often complement each other. The development and application of flexible informatics solutions that integrate the results of multiple research areas is of great significance. This study presents a multi-omics solution for large-scale analysis of MS data in metabolomics and proteomics data sets.
The Waters® omics research platform solution, including TransOmics informatics software, was used in conjunction with the Xevo G2-S QTof system for technical and biological replicate analysis.
Results and discussion
Performed metabolomics experiments included identification of low and high dose samples relative to control/QC samples. According to the experimental design, the sample should be divided into 3 different groups, and labeled ions are used for identification of different groups. The TransOmics (TOIML) process for metabolomics and lipidomics includes the following steps:
1. Import the original MS E continuous data set (six technical duplicates/groups)
2. Peak alignment to correct retention time offset between different analysis runs
3. Chromatographic peaks normalized for comparison between different sample runs
4. Chromatographic peak detection (peak selection)
5. Ion deconvolution, grouping ions by compound
6. Compound identification using existing custom databases
7. Perform data analysis to identify ions (characteristics) used to classify compounds into QC (QC), blank (matrix), and analyte (high dose)
The matrix background included a systematic evaluation matrix in which different analgesic standard mixtures A were added to obtain low doses (QC and high dose (blank) samples. QC samples were mixed by equal amounts of low and high dose samples And made.
The ACQUITY UPLC I-Class system was combined with the Xevo G2-S QTof to separate and analyze metabolites in positive electrospray mode with mass resolutions greater than 30k FWHM. Data is acquired in UPLC/MS E mode, an unsupervised acquisition method in which the mass spectrometer switches between low energy and high energy when performing alternate scans. Processing, searching and quantification using TOIML and specialized compound databases.
Steps 1, 2 and 3 of the TOIML process are described in detail elsewhere (TransOmics informatics software is supported by Nonlinear Dynamics). Prior to identification, the detected ions were grouped by principal component analysis, as shown in Figure 1, which shows a composite score map and a load map. It can be seen that the ions are mainly concentrated at the technical repetition level, and the sample achieves a clear separation.
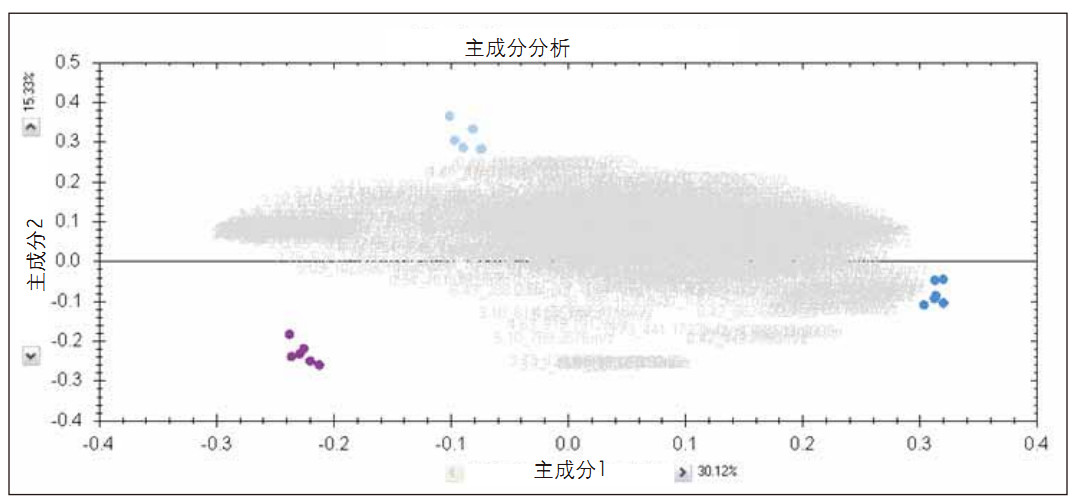
Figure 1. Principal component analysis of analytes (analgesic standard mixture A high dose; purple), blank (system evaluation matrix; light blue) and QC (quality control sample; dark blue)
Next, an integrated search tool was used to identify compounds to correctly identify the standards that could be detected in positive ion electrospray mode in a mixture of four analgesic standards. Figure 2 shows an overview of the TOIML compound search results page highlighting the identification of caffeine based on exact mass, retention time (optional), and theoretical isotope pattern distribution.
In addition to the previously described PCA, TOIML incorporates other multivariate statistical tools, including correlation and trend analysis. Figure 3 is an example showing a normalized trend plot of four spiked standards showing good agreement between the six technical replicates of each standard and the relative abundance consistent with the experimental design.
In addition, TOIML allows scientists to correlate analytical results with other omics data or to provide input data for independent statistical software packages such as EZinfo. The results of downstream bioinformatics (ie Umetrics software) can be re-introduced into analytical experiments to combine all compound data into a single table for review or sharing.
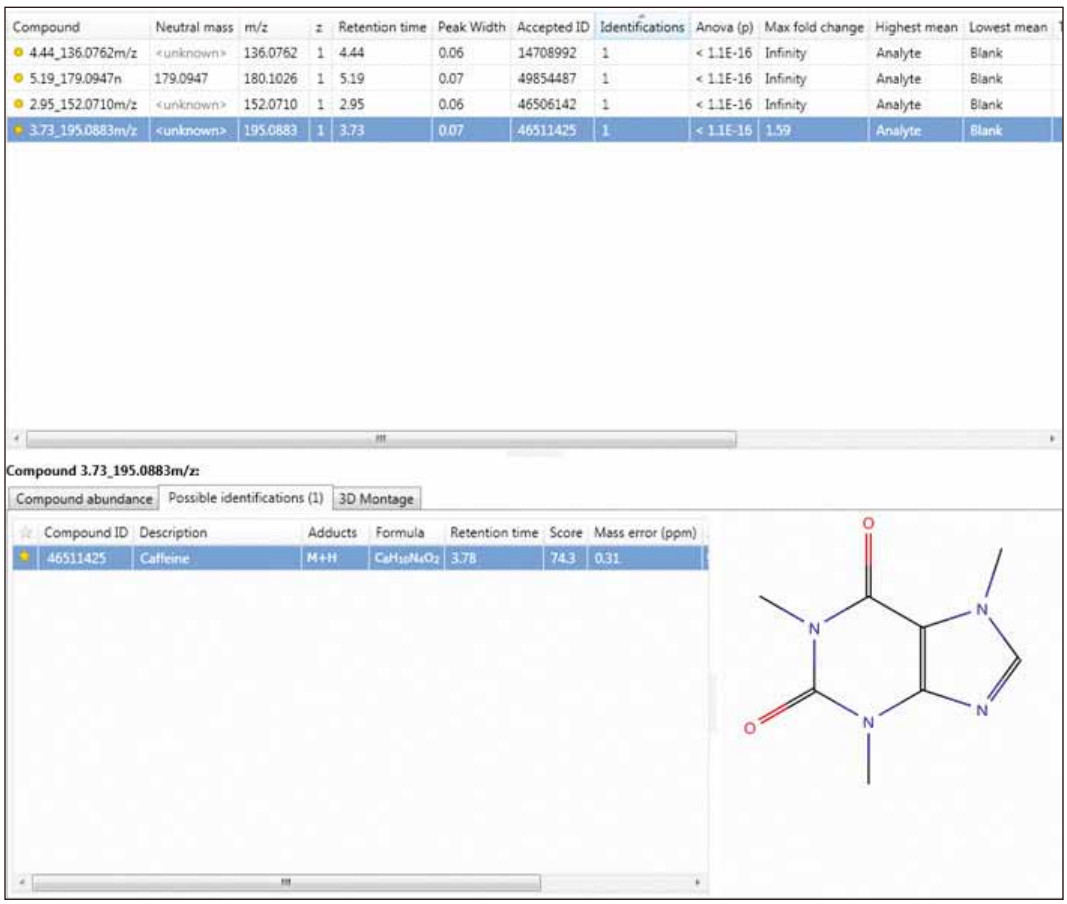
Figure 2. TOIML Compound Identification page.
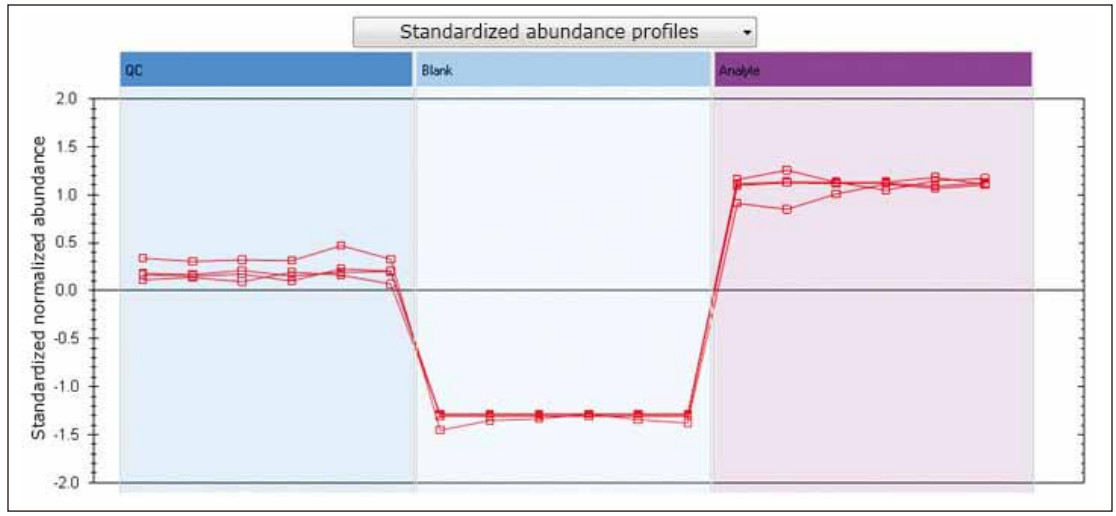
Figure 3. Normalized abundance analysis of analgesic standards.
In a proteomics experiment, three replicates of two 10 ng E. coli samples were analyzed, adding bovine serum albumin (BSA), alcohol dehydrogenase (ADH), enolase and glycogen phosphorylase. B. The on-column injection volume of the spiked protein in the first sample (mixture 1) was 1 femole, while the spiked protein in the second sample (mixture 2) was injected on the column 8,1, respectively. 2 and 0.5 fly moles.
Therefore, the nominal expected ratio (Mix 2: Mix 1) should be 8:1, 1:1, 2:1, and 0.5:1. In this study, peptides were separated and analyzed in LC/MS E acquisition mode using the nanoAC-QUITY UPLC system in conjunction with the Xevo G2-S QTof. Processing, searching and quantification were performed using TransOmics (TOIP) for proteomics and a species-specific database containing spiked protein sequence information.
The TOIP process includes the following steps:
1. Import the original MS E continuous data set (three technical replicates per sample)
2. Peak alignment to correct retention time offset between different analysis runs
3. Chromatographic peaks normalized for comparison between different sample runs
4. Chromatographic peak detection (peak selection)
5. Identify proteins and peptides using an integrated database search algorithm
6. Multivariate statistical analysis
7. Absolute and relative quantification
TOIP provides the same multivariate analysis tools as TOIML. Figure 4 shows a PCA example of the detected features, namely the set of charge states. It is apparent that the features are mainly concentrated at the level of technical duplication. The qualitative identification results of one of the spiked protein digests are shown in Figure 5. The normalized expression profiles of all peptides identified in this protein are shown in Figure 6. The latter's quantitative accuracy type was confirmed. This type can be obtained by label-free MS studies and LC/MS E- based acquisition strategies.
Figure 5 shows an overview of the qualitative results of LC/MS E acquisition of one analyte in a differential spiked sample. In this example, the on-column injection volume of BAS was 8 fmol, while the amount of E. coli digest was 10 ng. The results are shown in Figure 6, showing the relative relative quantification results.
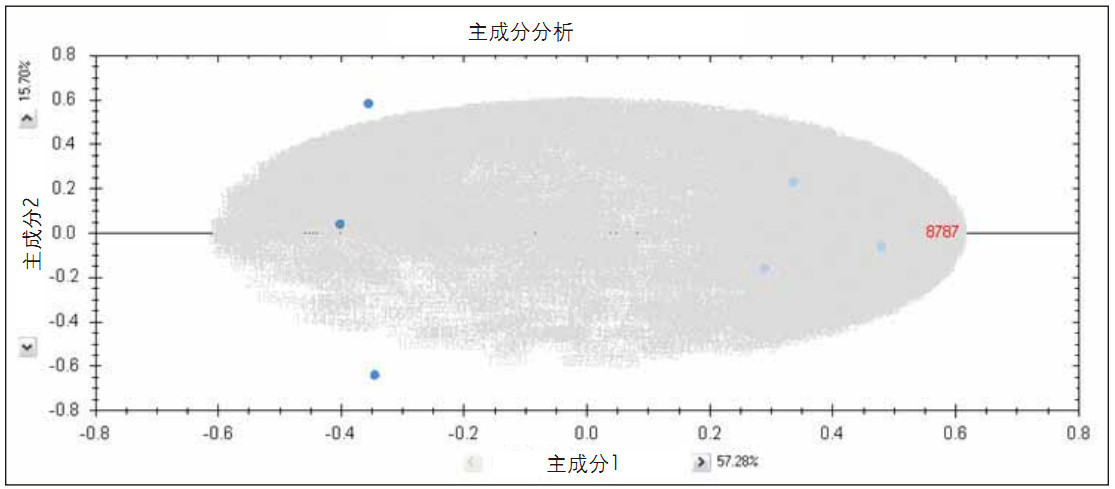
Figure 4. PCA plot of the characteristics (charge state group) of mixture 1 (dark blue) and mixture 2 (light blue) added to E. coli.
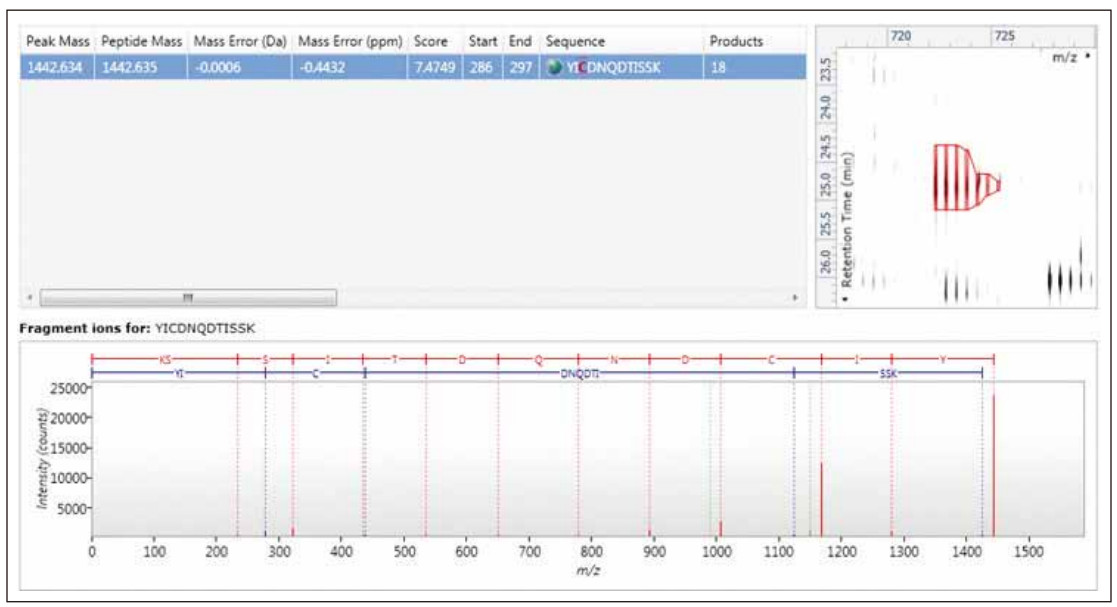
Figure 5. Qualitative LC/MS E identification of different concentrations of bovine serum albumin peptide added to E. coli. The clockwise display is followed by identification of relevant indicators (scores and errors), specific contour plots, and labeled product ion spectra.
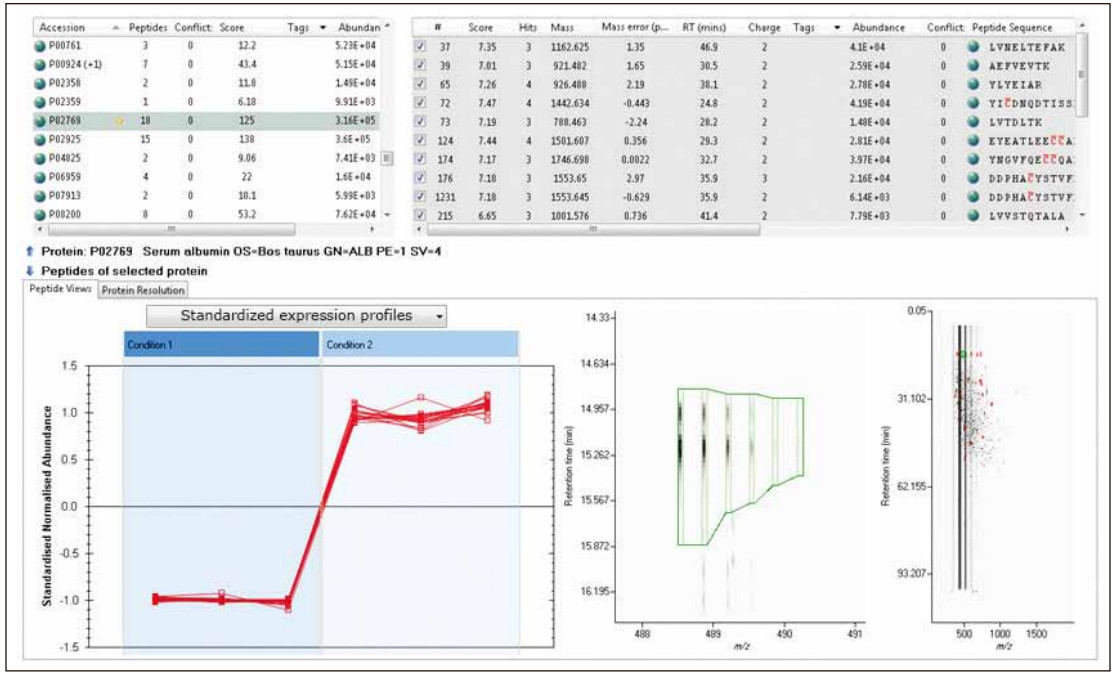
Figure 6. Quantitative analysis of peptides identified in bovine serum albumin.
Conclusion ■TransOmics informatics software provides an easy-to-use, scalable system for multi-omics research. UPLC/MS E (LC combined with data-independent acquisition of MS) ​​provides comprehensive qualitative and quantitative data sets in a single experiment. ■Quick access to supplemental information and association through metabolite, lipid and protein analysis
To download a clear application note, please click: http://?lid=134747751&cid=511436
Grow lighting can be one of the most essential factors to successful plant production in a commercial greenhouse because grow lights help to lighten shady spots and propagation areas in the greenhouse. If you are growing tropical plants, you may need to set up grow lights if the plants don't get at least eight hours of sun each day. Most crops benefit from supplemental lighting, when increased growth and quality is converted into added revenue. We provide Growing Light and complete systems for your greenhouses.
Greenhouse Growing Light,Greenhouse Grow Lights,Plant Grow Lights,Greenhouse Plant Lights
JIANGSU SKYPLAN GREENHOUSE TECHNOLOGY CO.,LTD , https://www.alibabagreenhouse.com